

Proteins are one of the most important molecules of life; the basic components of the genetic code. They have diverse functions such as catalyzing specific reactions, transporting and storing molecules, mechanical structure and support, signalling and motion. They fold into a unique 3 dimensional shape which allows them to perform unique and specific roles. If evolution is true, then it must be able to effectively evolve proteins. In part 1 I looked at the concept of protein sequence space and how experimental results have shown that functional and folding proteins are rare compared to non-functional and non folding proteins. In part 2 I look at theoretical, experimental and computational evidence in greater depth which shows that functional proteins are rare in total protein sequence space.
Theoretical and computational results for the rarity of functional proteins
Measuring the functional sequence complexity of proteins
Kirk Durston used actual data of known protein families to calculate the “functional complexity or functional information required to code for a particular protein family”. He calculated the ratio of non-functional proteins to functional proteins for a large number of different proteins and his results are consistent with other experimental results – namely that functional proteins are extremely rare in protein sequence space.

Figure 1: Number of non-functional proteins per functional proteins[i]
The results indicate that functional protein sequences are rare in total protein sequence space. For example the Ubiquitin protein sequence space consists of 10⁵² non-functional proteins per functional proteins. The red line indicates the total number of individual organism that have ever lived – it sets an extreme upper limit on the total sequences that could be searched by all organisms.
“Let us assume that every single gene in this total of 10³⁴ is unique and that evolution has been working on these genes for 4 Gyr completely changing each gene to some other unique, new gene every single year. This gives an extreme upper limit of 4 ×10⁴³ different amino acid sequences explored since the origin of life. The contribution to this number of sequences by viral and eukaryotic genomes is difficult to estimate but it is very unlikely to be orders of magnitude greater than the 4 ×10⁴³ sequences from bacteria”[ii]
Therefore any protein sequence space above the red line would be impossible for a blind random search to find given the total history of life.
Thoroughly sampling sequence space: Large-scale protein design of structural ensembles[iii]
The study looked at the distribution of different protein structures in sequence space. It looked at proteins with a residue length of 100 amino acids. The total number of structures they looked at was 253. They designed 100 structural variants for each structure by altering the dihedral angles of the protein structure backbone. From that they were then able to infer the residue entropy of each position.
“Designing to an ensemble of slight structural variants of the target structure produces a large diversity of high quality sequences, allowing for the exploration of a much broader range of sequence space than previous studies, and leading to novel insights into the determinants of protein sequence space.”
The residue entropy tells you how many different types of amino acids out of 20 can be substituted in that specific position without losing and destabilizing the structure. If the residue entropy is 5 it tells you that only 5 specific types of amino acids can be tolerated in that position. If it is 20 it means any amino acid in that position can be tolerated. Once the residue entropy for each position is obtained it is averaged to obtain the sequence entropy for the entire protein structure.

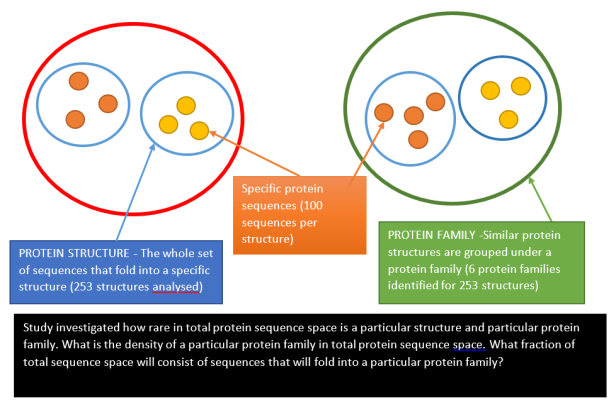
The study then grouped all 253 structures into 6 protein families and measured the sequence entropy distribution. For example in the toxin protein family, the most frequent sequence entropy is 5.8 with 30% of all proteins in that family possessing that sequence entropy. From sequence entropy you can calculate the protein density for that structure in total sequence space.

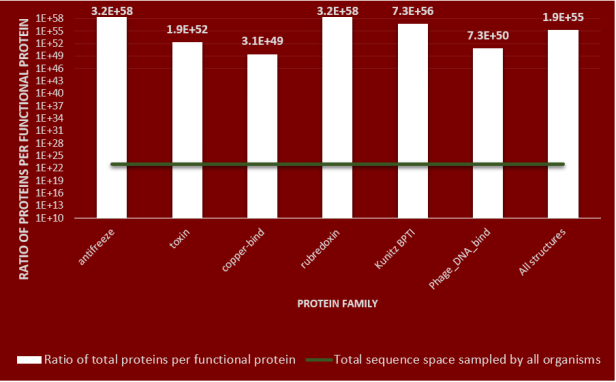
Figure 3: Density of protein families in protein sequence space
The figure shows the density of functional proteins in protein sequence space. For the copper-bind protein family structure – the density is 10⁴⁹ proteins per one functional protein. It means in order to find a single folding and functional protein sequence that will fold into the copper-bind structure you will have to search 10⁴⁹ sequences. The average protein density for all 253 structures is 10⁵⁵. There is one folding protein per 10⁵⁵ proteins.
The total sequence space that has been sampled by bacterial organisms (which have the largest effective population sizes) since life originated on earth is indicated by the green line as 10²³. Evolution has only sampled a small fraction of the sequence space needed to find a folding functional protein. These results are again consistent with other results which have used different methods to converge on the same conclusion- sequence space is largely populated by nonfunctional, non-folding proteins.

Protein topology and stability define the space of allowed sequences[iv]
Scientists Koel and Levitt used three different methods to estimate the folding sequence space for 10 different proteins ranging in size from 56 to 310 residues. They determined what fraction of the total protein sequence space will fold into stable structures.
“We compared these 3 independent multiple sequence alignments for 10 different proteins, ranging in size from 56 to 310 residues. We observed that the subset of the sequence space derived by using our design procedure is similar in size to the sequence spaces observed in nature. These results suggest that the volume of sequence space compatible with a given protein fold is defined by the length of the protein as well as by the topology (i.e., geometry of the polypeptide chain) and the stability (i.e., free energy of denaturation) of the fold.”
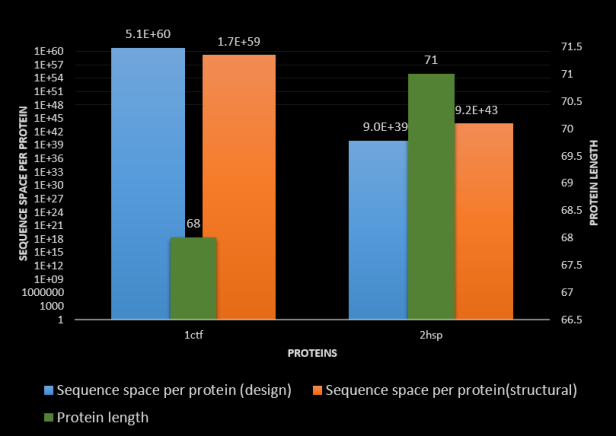
The figure shows the results they obtained for two proteins. 1ctf and 2 hsp which are 68 and 71 residues long respectively. Their results show the sequence space obtained for 2 different methods. For example – the 2hsp protein the sequence spacer per folding protein is 10³⁹ based on the structural method. This means there is one folding protein per 10³⁹ protein sequences.
They also state an important factor that determines the sequence space: “…independent of functional fitness, it is the topology of a protein, its length, and its stability that define the size of the sequence space that is compatible with its structure”. This means the topology, structure, stability and length largely determine the sequence space of folding proteins that may possess a biochemical function.
In conclusion both experimental and theoretical results consistently indicate that functional proteins are extremely rare in protein sequence space. The values differ depending on the complexity and type of protein structure.
Why is it a problem?
- Firstly it poses a problem for evolutionary accounts of how life originated. Finding a functional protein by chance is highly unlikely.
- It poses a problem in that random mutations must search through a vast space of nonfunctional proteins before finding a functional one which natural selection can then act on.
Bacterial populations have the highest effective population sizes and shortest generation periods. Which means they are a generous overestimation what resources evolution has at its disposable. The whole history of bacterial organisms would not have sampled enough of sequence space to find a folding stable functional protein. The following calculation illustrates the problem:

- Organisms have only sampled an extremely small fraction of the total available protein sequence space – well below what is required to find a functional protein.
References
[i] Kirk K Durston, David KY Chiu, David L Abel and Jack T Trevors. Measuring the functional sequence complexity of proteins. Theoretical Biology and Medical Modelling 2007, 4:47 doi:10.1186/1742-4682-4-47
[ii] David T. F. Dryden, Andrew R. Thomson and John H. White. How much of protein sequence space has been explored by life on Earth? J. R. Soc. Interface (2008) 5, 953–956. doi:10.1098/rsif.2008.0085
[iii] STEFAN M. LARSON, JEREMY L. ENGLAND, JOHN R. DESJARLAIS, AND VIJAY S. PANDE. Thoroughly sampling sequence space: Large-scale protein design of structural ensembles. Protein Science (2002), 11:2804–2813.
[iv] Patrice Koehl and Michael Levitt. Protein topology and stability define the space of allowed sequences. www.pnas.org/cgi/doi/10.1073/pnas.032405199
Axe DD. The case against a Darwinian origin of protein folds. BIO-Complexity 2010(1):1-12. doi:10.5048/BIO-C.2010.1
